Evaluate clustering goodness-of-fit
Introduction
In this example, we will show how to use scDesign3Py to evaluate the clustering goodness-of-fit for different cell-type assignments. If the true labels are unavailable and we have little prior knowledge, the scDesign3Py BIC can serve as an unsupervised metric.
Import packages and Read in data
import pacakges
import anndata as ad
import pandas as pd
import scanpy as sc
import scDesign3Py
from sklearn.metrics import adjusted_rand_score
Read in data
The raw data is from the R package DuoClustering2018 and converted to .h5ad file using the R package sceasy.
The cluster result is also got from the R package DuoClustering2018 as the package has already provided various clustering results.
data = ad.read_h5ad("data/Zhengmix4eq.h5ad")
cluster_res = pd.read_csv("data/clusterres_Zhengmix4eq.csv",index_col=0)
cluster_res = cluster_res[cluster_res["method"].isin(["PCAKmeans"]) & (cluster_res["run"] == 1)]
For demonstration purpose, we use the Zhengmix4eq dataset in the package with top 30 highly variable genes and the corresponding k-means clustering results with \(k = 2 \sim 10\).
kmeans_res = cluster_res.groupby("k")
ncell = data.n_obs
ngene = 30
ntrain = int(ncell/5)
# choose HVG genes
data.layers["log"] = data.X.copy()
sc.pp.normalize_total(data,target_sum=1e4,layer="log")
sc.pp.log1p(data,layer="log")
sc.pp.highly_variable_genes(data,layer="log",n_top_genes=ngene)
data = data[:,data.var["highly_variable"] == True]
train_data = data[data.obs.sample(n=ntrain,random_state=123).index,:]
train_data
View of AnnData object with n_obs × n_vars = 711 × 30
obs: 'barcode', 'phenoid', 'total_features', 'log10_total_features', 'total_counts', 'log10_total_counts', 'pct_counts_top_50_features', 'pct_counts_top_100_features', 'pct_counts_top_200_features', 'pct_counts_top_500_features', 'sizeFactor', 'cell_type'
var: 'id', 'symbol', 'mean_counts', 'log10_mean_counts', 'rank_counts', 'n_cells_counts', 'pct_dropout_counts', 'total_counts', 'log10_total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'log1p', 'hvg'
obsm: 'X_pca', 'X_tsne'
layers: 'logcounts', 'normcounts', 'log'
Simulation
We then use different cell-type clustering information to simulate new data.
simu_res = {}
for key,value in kmeans_res:
print(f"K number: {key} fitting.")
tmp = value[["cell","cluster"]]
tmp.index = tmp["cell"]
train_data.obs["cell_type"] = tmp.loc[train_data.obs.index,"cluster"].tolist()
train_data.obs["cell_type"] = train_data.obs["cell_type"].astype("int").astype("str").astype("category")
test = scDesign3Py.scDesign3(n_cores=2, parallelization="pbmcmapply")
test.set_r_random_seed(123)
res = test.scdesign3(anndata=train_data,
celltype = 'cell_type',
corr_formula = "1",
mu_formula = "cell_type",
sigma_formula = "cell_type",
copula = "gaussian",
default_assay_name = "counts",
family_use = "nb",
usebam = False)
simu_res[key] = res
Visualization
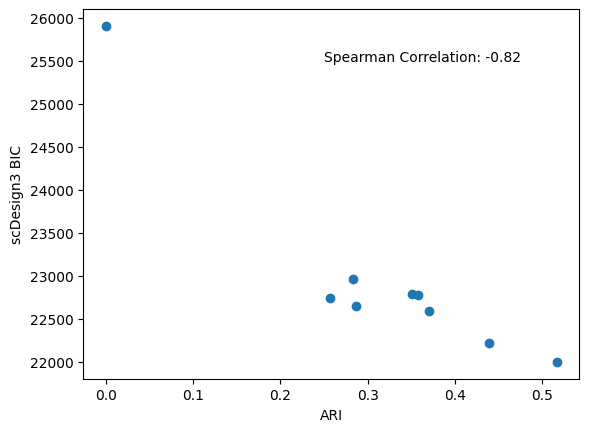
After the simulations, we can check the BIC provided by our package and the calculated ARI to evaluate k-means clustering qualities.
import matplotlib.pyplot as plt
Show code cell source
bics = pd.Series([v["model_bic"]["bic.marginal"] for v in simu_res.values()])
ari = pd.Series([adjusted_rand_score(v["cluster"],v["trueclass"]) for _,v in kmeans_res])
spearman_corr = bics.corr(ari,method="spearman")
# plot
plt.scatter(x=ari,y=bics)
plt.xlabel('ARI')
plt.ylabel('scDesign3 BIC')
plt.text(x=0.25,y=25500,s="Spearman Correlation: %.2f" % spearman_corr)
plt.show()