Simulate datasets with batch effect
Introduction
In this example, we will show how to use scDesign3Py to simulate data with original batch effects and how to remove the batch effects. We will also demostrate how to add ariticial batch effects.
Import packages and Read in data
import pacakges
import copy
import anndata as ad
import numpy as np
import scDesign3Py
Read in data
The raw data is from the SeuratData package. The data is called pbmcsca in the package; it is PBMC Systematic Comparative Analysis dataset from the Broad Institute. The raw data is converted to .h5ad file using the R package sceasy.
To save time, we only choose the top 30 genes.
data = ad.read_h5ad("data/BATCH.h5ad")
data = data[:,0:30]
data.layers["log"] = np.log1p(data.X)
data
AnnData object with n_obs × n_vars = 6276 × 30
obs: 'phenoid', 'orig.ident', 'nCount_RNA', 'nFeature_RNA', 'nGene', 'nUMI', 'percent.mito', 'Cluster', 'Experiment', 'Method', 'ident', 'batch', 'cell_type'
var: 'name'
layers: 'log'
The column batch in this example dataset’s obs contains the batch information.
data.obs["batch"].head()
pbmc2_10X_V2_AAACCTGAGATGGGTC 10x Chromium (v2)
pbmc2_10X_V2_AAACCTGAGCGTAATA 10x Chromium (v2)
pbmc2_10X_V2_AAACCTGAGCTAGGCA 10x Chromium (v2)
pbmc2_10X_V2_AAACCTGAGGGTCTCC 10x Chromium (v2)
pbmc2_10X_V2_AAACCTGGTCCGAACC 10x Chromium (v2)
Name: batch, dtype: category
Categories (2, object): ['10x Chromium (v2)', '10x Chromium (v3)']
Simulation
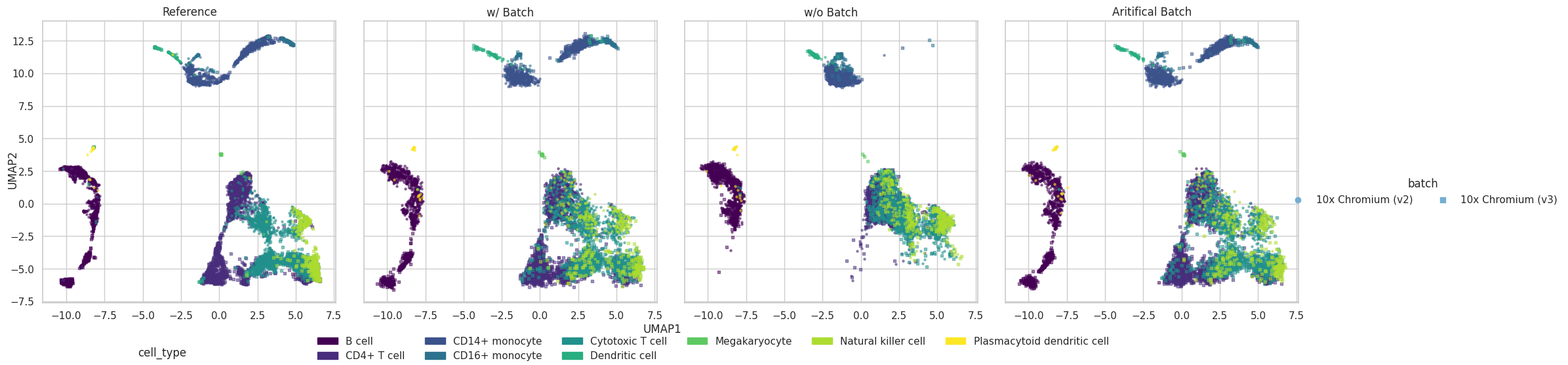
We can simulate a new data with batch effect information.
test = scDesign3Py.scDesign3(n_cores=3,parallelization="pbmcmapply")
test.set_r_random_seed(123)
simu_res = test.scdesign3(anndata=data,
default_assay_name = "counts",
celltype = "cell_type",
other_covariates = "batch",
mu_formula = "cell_type + batch",
sigma_formula = "1",
family_use = "nb",
usebam = False,
corr_formula = "1",
copula = "gaussian",)
simu_count = simu_res["new_count"]
We can also remove the batch effect and generate new data.
# create instance
batch = scDesign3Py.scDesign3(n_cores=3,parallelization="pbmcmapply")
# construct data
batch_data = batch.construct_data(
anndata=data,
default_assay_name="counts",
celltype = "cell_type",
other_covariates = "batch",
corr_formula = "1"
)
# fit marginal
batch_marginal = batch.fit_marginal(
mu_formula="cell_type + batch",
sigma_formula="1",
family_use="nb",
usebam=False,
)
# fit copula
batch_copula = batch.fit_copula()
In here, we remove the batch effect by setting its coefficient to zero for all genes’ marginal fits. Then, we use the new sets of coefficients to generate the parameters for all genes across all cells.
batch_null = scDesign3Py.scDesign3(n_cores=3,parallelization="pbmcmapply")
batch_data_null = batch_null.construct_data(
anndata=data,
default_assay_name="counts",
celltype = "cell_type",
other_covariates = "batch",
corr_formula = "1"
)
batch_marginal_null = copy.deepcopy(batch_marginal)
for k,_ in batch_marginal_null.items():
batch_marginal_null.rx2(k).rx2("fit").rx2("coefficients")[-1] = 0
batch_para_null = batch_null.extract_para(
marginal_dict=batch_marginal_null,
family_use="nb",
)
batch_null.set_r_random_seed(123)
batch_null.copula = "gaussian"
batch_new_count_null = batch_null.simu_new(
copula_dict=batch_copula["copula_list"],
family_use="nb",
important_feature=batch_copula["important_feature"],
)
Note
Here, as we direct use another copula model result, to tell the scDesign3Py how to change the copula dict back to R list, the class property copula should be specified.
Additionally, we can alter the batch effect information by mannually change the estimated coefficient for batch effect in each gene’s marginal model. Then, we can simulate new dataset with altered batch effect information.
batch_alter = scDesign3Py.scDesign3(n_cores=3,parallelization="pbmcmapply")
batch_data_alter = batch_alter.construct_data(
anndata=data,
default_assay_name="counts",
celltype = "cell_type",
other_covariates = "batch",
corr_formula = "1"
)
batch_marginal_alter = copy.deepcopy(batch_marginal)
for k,_ in batch_marginal_alter.items():
batch_marginal_null.rx2(k).rx2("fit").rx2("coefficients")[-1] = np.random.normal(loc=1,scale=2)
batch_para_alter = batch_alter.extract_para(
marginal_dict=batch_marginal_alter,
family_use="nb",
)
batch_alter.set_r_random_seed(123)
batch_alter.copula = "gaussian"
batch_new_count_alter = batch_alter.simu_new(
copula_dict=batch_copula["copula_list"],
family_use="nb",
important_feature=batch_copula["important_feature"],
)
We then create the corresponding anndata.AnnData object.
simu_anndata_list = []
for count_mat in [simu_count,batch_new_count_null,batch_new_count_alter]:
tmp = ad.AnnData(X=count_mat,obs=batch_data["newCovariate"])
tmp.layers["log"] = np.log1p(tmp.X)
simu_anndata_list.append(tmp)
Visulization
plot = scDesign3Py.plot_reduceddim(
ref_anndata=data,
anndata_list=simu_anndata_list,
name_list=["Reference", "w/ Batch", "w/o Batch", "Aritifical Batch"],
assay_use="log",
color_by = "cell_type",
shape_by = "batch",
n_pc=20,
point_size=5,
)
UMAP
plot["p_umap"]