Simulate multi-omics data from single-omic data
Introduction
In this example, we show how to use scDesign3Py to simulate multi-omics (RNA expression + DNA methylation) data by learning from real data that only have a single modality. The example data and aligned low-dimensional embeddings are from Pamona.
Import packages and Read in data
import pacakges
import anndata as ad
import pandas as pd
import scDesign3Py
import scanpy as sc
Read in the reference data
meth_exp = ad.read_h5ad("data/SCGEMMETH.h5ad")
rna_exp = ad.read_h5ad("data/SCGEMRNA.h5ad")
Simulation
We first use the step-by-step functions to fit genes’ marginal models, copulas, and extract simulation parameters separately for the scRNA-seq data and the DNA methylation data.
rna = scDesign3Py.scDesign3(n_cores=1,parallelization="mcmapply")
rna_data = rna.construct_data(
anndata=rna_exp,
default_assay_name="logcounts",
celltype="cell_type",
spatial=["UMAP1_integrated","UMAP2_integrated"],
corr_formula="1"
)
meth = scDesign3Py.scDesign3(n_cores=1,parallelization="mcmapply")
meth_data = meth.construct_data(
anndata=meth_exp,
default_assay_name="counts",
celltype="cell_type",
spatial=["UMAP1_integrated", "UMAP2_integrated"],
corr_formula="1",
)
Note here we actually treat the 2D aligned UMAPs as a kind of “pseudo”-spatial data. We use the tensor regression spline to fit two ref datasets seperately.
rna.set_r_random_seed(123)
rna_marginal = rna.fit_marginal(
mu_formula="te(UMAP1_integrated, UMAP2_integrated, bs = 'cr', k = 10)",
sigma_formula="te(UMAP1_integrated, UMAP2_integrated, bs = 'cr', k = 5)",
family_use="gaussian",
usebam=False,
)
meth.set_r_random_seed(123)
meth_marginal = meth.fit_marginal(
mu_formula="te(UMAP1_integrated, UMAP2_integrated, bs = 'cr', k = 10)",
sigma_formula="1",
family_use="binomial",
usebam=False,
)
rna_copula = rna.fit_copula(copula="vine")
meth_copula = meth.fit_copula(copula="vine")
rna_para = rna.extract_para(new_covariate=pd.concat([rna_data["dat"], meth_data["dat"]], axis=0))
meth_para = meth.extract_para(new_covariate=pd.concat([rna_data["dat"], meth_data["dat"]], axis=0))
Then, we combined the cell covariates from both the scRNA-seq data and the DNA methylation data as the new covariate to simulate the two new datasets with parameters from scRNA-seq data and the DNA methylation data separately.
rna_res = rna.simu_new(new_covariate=pd.concat([rna_data["dat"], meth_data["dat"]], axis=0),
important_feature=rna_copula["important_feature"],)
meth_res = meth.simu_new(new_covariate=pd.concat([rna_data["dat"], meth_data["dat"]], axis=0),
important_feature=meth_copula["important_feature"],)
Visualization
We combine the two synthetic datasets and obtain the UMAP embeddings for the combined dataset.
count_combine = pd.concat([rna_res,meth_res], axis=1)
combine_exp = ad.AnnData(X=count_combine)
sc.tl.pca(combine_exp, n_comps=5)
sc.pp.neighbors(combine_exp, n_neighbors=30, n_pcs=5)
sc.tl.umap(combine_exp, min_dist=0.7)
rna_exp.obsm["cov"] = rna_exp.obs.iloc[:,0:5]
sc.pp.neighbors(rna_exp, n_neighbors=30, use_rep="cov")
sc.tl.umap(rna_exp, min_dist=0.7)
meth_exp.obsm["cov"] = meth_exp.obs.iloc[:,0:5]
sc.pp.neighbors(meth_exp, n_neighbors=30, use_rep="cov")
sc.tl.umap(meth_exp, min_dist=0.7)
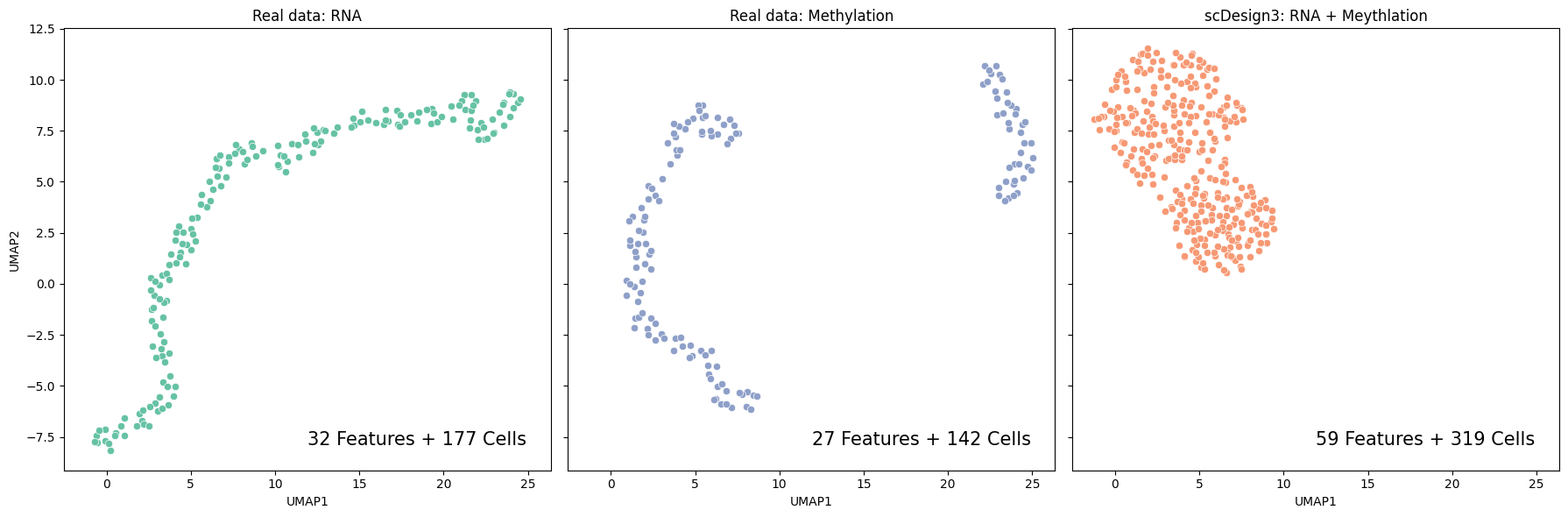
Then we visualize the UMAP embeddings for the inputted scRNA-seq data, DNA methylation data, and the combined synthetic data.
import matplotlib.pyplot as plt
import seaborn as sns
Show code cell source
fig, axs = plt.subplots(1, 3, figsize=(18, 6), sharex=True, sharey=True)
sns.scatterplot(data=pd.DataFrame(rna_exp.obsm["X_umap"],columns=["UMAP1","UMAP2"]), x="UMAP1", y="UMAP2", ax=axs[0], color = "#66c2a5")
axs[0].set_title("Real data: RNA")
axs[0].text(0.95, 0.05, "32 Features + 177 Cells", verticalalignment="bottom", horizontalalignment="right", transform=axs[0].transAxes, fontsize=15)
sns.scatterplot(data=pd.DataFrame(meth_exp.obsm["X_umap"],columns=["UMAP1","UMAP2"]), x="UMAP1", y="UMAP2", ax=axs[1], color = "#8ea0c9")
axs[1].set_title("Real data: Methylation")
axs[1].text(0.95, 0.05, "27 Features + 142 Cells", verticalalignment="bottom", horizontalalignment="right", transform=axs[1].transAxes, fontsize=15)
sns.scatterplot(data=pd.DataFrame(combine_exp.obsm["X_umap"],columns=["UMAP1","UMAP2"]), x="UMAP1", y="UMAP2", ax=axs[2], color = "#f79974")
axs[2].set_title("scDesign3: RNA + Meythlation")
axs[2].text(0.95, 0.05, "59 Features + 319 Cells", verticalalignment="bottom", horizontalalignment="right", transform=axs[2].transAxes, fontsize=15)
plt.tight_layout()
plt.show()